Table of Contents
Overview of Mechanistic Pathways of Organic Reactions:
Organic chemistry is fundamental to numerous scientific and industrial disciplines, playing a vital role in various domains such as pharmaceuticals, agriculture, materials research, and environmental studies. The core of organic chemistry focuses on the examination of reactions, which are the mechanisms by which molecules change structure or composition. Mechanistic Pathways of Organic Reactions guide the transformations by outlining the precise sequence of actions that transform reactants into products. Comprehending these routes is crucial for achieving proficiency in organic chemistry, as they not only unveil the progression of reactions but also elucidate the underlying reasons for their specific behavior.
Given the growing intricacy of organic reactions and the emergence of multidisciplinary areas, there is a pressing demand for extensive collections of reaction data on a wide scale. These databases function as a storage of information, documenting established reactions together with their accompanying molecular pathways. As the discipline advances, the size and range of these databases increase, propelled by improvements in computational chemistry, machine learning, and data science. These databases are crucial for forecasting reaction outcomes, building new synthetic routes, and discovering new mechanistic insights that could potentially lead to significant advancements in numerous industries.
This article explores the importance of extensive reaction datasets, specifically those that examine the mechanisms involved in organic reactions. We will examine the development of these datasets, including their elements, uses, difficulties, and prospects. Additionally, we will explain how artificial intelligence (AI) might improve their usefulness.
Analyzing the Mechanistic Pathways of Organic Reactions:
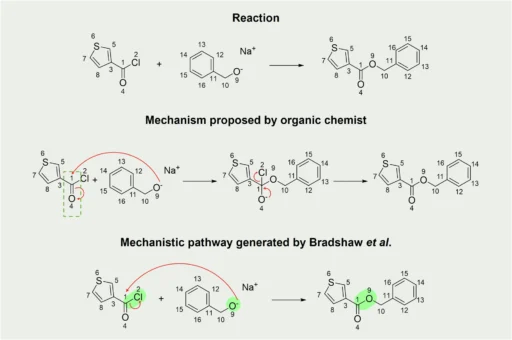
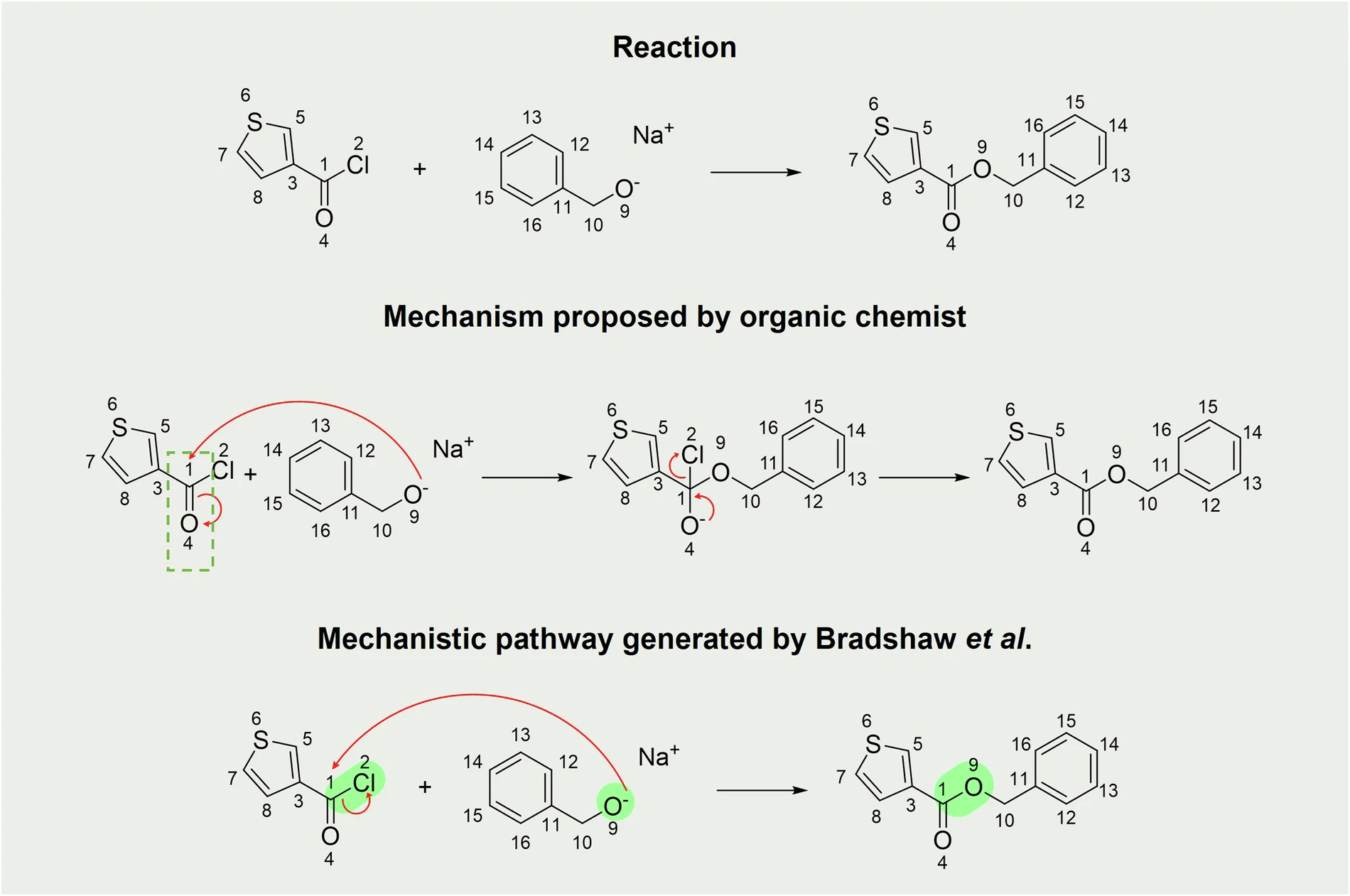
Mechanistic pathways of Organic Reactions are precise and thorough sequences of steps that define the progression of a chemical reaction from its initial reactants to the final products. Mechanistic Pathways of Organic Reactions are essential in organic chemistry because they serve as a guide for understanding the chemical transformation process. Each step within a Mechanistic Pathways of Organic Reactions entails a distinct sequence of occurrences, such as the formation or breaking of chemical bonds, the movement of electrons, and the generation or elimination of intermediate substances. Chemists can gain knowledge about Mechanistic Pathways of Organic Reactions, and a reaction’s energy demands, anticipate possible secondary reactions, and improve conditions to achieve higher yields or selectivity through the analysis of these processes.
Computational models frequently reinforce mechanistic routes, which are not just abstract concepts based on empirical evidence. They play a critical role in influencing a reaction’s outcome. For example, a reaction that occurs through a radical mechanism would exhibit distinct behavior under different conditions compared to a reaction that progresses through a coordinated process. Comprehending these distinctions can be critical in managing reaction results, making mechanistic pathways an essential tool in the chemist’s arsenal. Mechanistic Pathways of Organic Reactions:
The progression of reaction datasets in the field of chemistry is fascinating:
Documenting chemical reactions has a long and extensive history, dating back to the early stages of chemistry when alchemists meticulously documented their experiments. However, these records often displayed inconsistencies, lacked comprehensiveness, and were dispersed across numerous publications. As chemistry advanced, it became clear that there was a need for organized record-keeping, which resulted in the development of databases specifically for reactions.
Initially, chemical databases were relatively small and consisted of meticulously managed compilations of reactions obtained from published scientific literature. These databases frequently have restrictions on the types of reactions they cover or the specific areas of chemistry they focus on. However, the emergence of digital technology and the internet enabled the creation of expansive and all-encompassing databases that academics could access and search globally.
The integration of computational chemistry and machine learning into dataset generation and analysis initiated the true revolution. These technologies made it possible for response databases to grow quickly and include large amounts of data from many sources, such as the results of high-throughput experiments and computer predictions. Currently, extensive reaction datasets are essential resources in the field of chemistry, offering researchers the necessary information to plan experiments, anticipate reaction results, and investigate unexplored regions of chemical space.
Components of a Large-Scale Reaction Dataset:
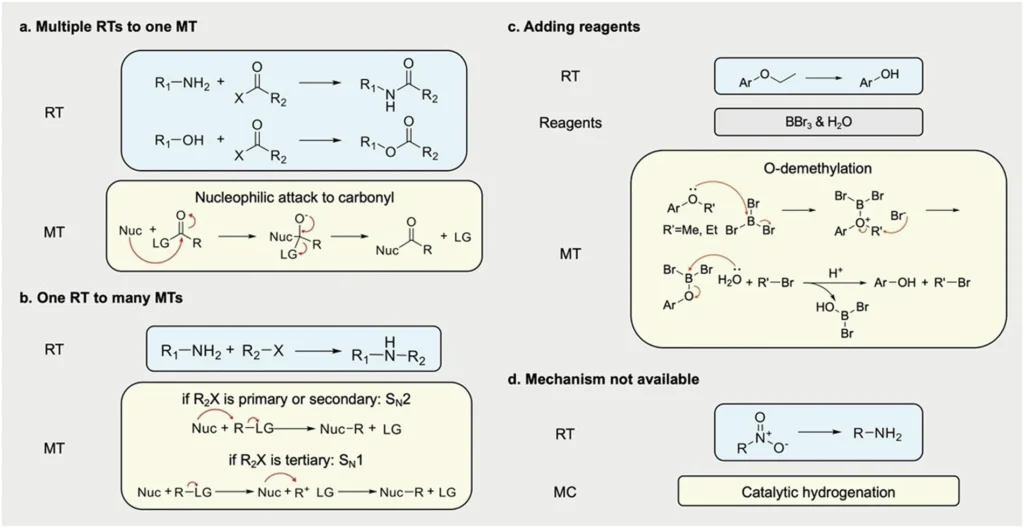
A comprehensive reaction dataset is not simply a compilation of reactions, but rather a sophisticated and multifaceted repository of information that requires meticulous curation and maintenance to ensure its precision and usefulness. The essential elements of such a dataset include the techniques used for data gathering, the specific types of data incorporated, the annotation of mechanical processes, and the uniformity of data formats. Mechanistic Pathways of Organic Reactions:
Data Collection: Sources and Methods
Compiling a comprehensive dataset for a large-scale reaction is a difficult endeavor that requires obtaining data from diverse sources. The sources encompass published research, experimental findings, and computational forecasts. Literature mining is a widely used technique for gathering data, in which algorithms analyze scholarly papers to extract pertinent details about reactions. Nevertheless, this approach necessitates meticulous validation to guarantee the accuracy and comprehensiveness of the retrieved data.
Experimental data is an important component of a response dataset. High-throughput screening assays frequently acquire this data, examining several reactions under various conditions to determine their results. This experimental data is extremely significant as it offers empirical proof of how reactions function under different circumstances.
Computational predictions are also crucial in generating extensive reaction databases. Algorithms that simulate the behavior of molecules and processes, employing established chemical principles, derive the predictions. While computational predictions may not always be as reliable as experimental data, they can provide valuable insights into reactions that remain empirically unexplored. Mechanistic Pathways of Organic Reactions:
Key Elements: Reactants, Products, and Intermediates
The fundamental components of each reaction dataset are the reactants, products, and intermediates that characterize each reaction. Reactants are the initial substances that undergo a chemical change during a reaction, while products are the outcomes of this process. However, the reaction mechanism forms intermediates, which are temporary entities that undergo further reactions. It is crucial to document these factors to comprehend the complete alteration that takes place throughout a reaction.
For each reaction, it is critical to include comprehensive information about the reactants and products in the dataset, including their chemical structures, molecular weights, and other relevant attributes. Documenting intermediates can be particularly difficult due to their transient nature and the challenges associated with isolating and characterizing them. Nevertheless, comprehending these intermediate stages is essential for clarifying the complete molecular pathway of a process.
Exploring the Process of Annotating Mechanistic Steps: Difficulties and Optimal Approaches
An annotation of mechanistic processes is an essential component of a comprehensive dataset for large-scale reactions. This procedure entails elucidating the precise sequence of events that transpire throughout a reaction, encompassing the formation and breaking of chemical bonds, the movement of electrons, and the creation of intermediate compounds. Precise annotation is critical for understanding a reaction’s progression and forecasting the results of analogous reactions.
Nevertheless, the process of annotating mechanistic processes is not devoid of difficulties. Annotating complex reactions, particularly those that include numerous stages or uncommon intermediates, can present challenges in terms of accuracy. Occasionally, the precise mechanism may be unclear or may encompass other conflicting paths. To address these issues, we recommend utilizing a blend of experimental data, computational modeling, and expert knowledge during the annotation process. This approach ensures that the mechanistic annotations are both accurate and comprehensive.
Data Standardization and Format:
Data standardization refers to the process of establishing a uniform format for data. This involves ensuring that data is organized and structured consistently, making it easier to analyze and interpret. Standardizing data helps to eliminate inconsistencies and discrepancies, allowing for more accurate and reliable analysis.
Data standardization is an essential element of a large-scale comprehensive reaction dataset. Different forms, such as SMILES (Simplified Molecular Input Line Entry System), InChI (International Chemical Identifier), and reaction SMIRKS (SMILES Reaction String), can express chemical reactions. Ensuring uniformity throughout a dataset is critical for its usability, as it facilitates improved cross-referencing between diverse datasets and guarantees global access to the information for researchers.
Data standardization means creating a consistent way to describe processes, including the makeup of reactants and products, the conditions under which the reaction happens, and the labeling of the steps that make the reaction happen. Standardization is crucial to facilitate the seamless sharing and retrieval of data among researchers across various disciplines and institutions.
Utilization of Extensive Reaction Datasets:
Extensive reaction databases have a variety of applications in organic chemistry and other fields. Predictive modeling, drug discovery, synthesis planning, and cross-referencing with experimental data are just a few of the uses for these datasets. The subsequent sections delve into some of the most notable applications of these datasets.
Enhanced Predictive Modeling in Organic Chemistry:
Predictive modeling is a major area where large-scale reaction datasets are highly valuable. Predictive modeling is the process of utilizing available data to forecast the results of untried reactions. This skill is especially advantageous in the field of organic chemistry since it allows for the anticipation of reaction results, leading to time and resource savings in the laboratory.
Machine learning algorithms construct predictive models by scrutinizing extensive databases of established behavior to discern patterns and correlations. We can then use these models to predict the outcomes of new reactions, even ones that have not yet undergone experimental testing. The ability to anticipate reaction outcomes is extremely useful for chemists involved in the development of novel synthetic pathways or the enhancement of existing ones.
For instance, predictive models can assist chemists in identifying the most probable outcomes of a reaction, as well as determining the optimal reaction conditions and predicting potential secondary reactions. We can use this data to create more precise and effective synthetic pathways, which reduces the need for laboratory trial-and-error experimentation.
This facilitates the process of discovering and designing drugs:
Extensive reaction databases are vital in the domain of drug discovery and design. Drug discovery researchers frequently face the challenge of manufacturing novel molecules that have not been previously synthesized. To achieve a desired therapeutic outcome, researchers must intentionally formulate these chemicals to interact with specific biological targets.
Reaction databases are useful for finding possible ways to make new drug candidates, guessing how these molecules will act in different situations, and finding the best reaction settings to get the highest yields and selectivity. These datasets have the potential to greatly expedite the drug discovery process by granting researchers access to extensive reaction data, thereby reducing the time and costs associated with introducing new pharmaceuticals to the market.
Furthermore, researchers can use reaction databases to predict the behavior of novel drug candidates in biological systems. Through the analysis of data obtained from prior trials, researchers can discern patterns and associations that may indicate the potential interactions between a novel molecule and its intended target. Researchers can utilize this data to develop more potent and targeted medications, thereby enhancing their likelihood of achieving favorable outcomes in clinical trials. Mechanistic Pathways of Organic Reactions:
Enhancing the processes of synthesis, planning, and optimization:
Synthesis planning is a crucial component of organic chemistry that entails devising a series of reactions to create a desired product from a given set of initial substances. Large-scale reaction datasets are critical for synthesis planning because they provide chemists with access to extensive information about known reactions and their results.
Chemists can determine the most optimal methods to create intricate molecules by examining a vast assortment of established reactions. This approach not only saves time but also has the potential to decrease expenses and minimize the negative effects on the environment. We can also utilize reaction datasets to optimize reaction parameters such as temperature, pressure, and solvent selection, thereby enhancing yields and selectivity.
For example, when tasked with creating a new molecule, a chemist can use a collection of reactions to identify similar reactions from previous experiments. Through analysis of the conditions and outcomes of these reactions, the chemist can devise a synthetic pathway that is more likely to achieve success. This strategy has the potential to greatly diminish the necessity for trial-and-error testing in the laboratory, resulting in time and resource savings.
Utilizing Experimental Data for Cross-Referencing:
Another notable use of extensive response datasets is their integration with experimental data for cross-referencing purposes. Empirical data provides tangible proof of how reactions function in specific circumstances, and comparing this data with other reaction datasets can help verify forecasts and discover novel patterns.
For instance, when a predictive model proposes a certain result for a reaction, verifying this forecast by comparing it with experimental data helps validate its precision. If the forecast aligns with the empirical findings, it might instill trust in the model’s dependability. On the other hand, if the forecast does not align with the experimental data, it can stimulate additional inquiry to comprehend the difference and enhance the model.
Cross-referencing with experimental data is also advantageous for discerning novel patterns in reaction behavior. Through the analysis of extensive datasets of experimental findings, researchers can discern patterns and relationships that may not be readily evident from individual trials. These trends offer useful insights into the parameters that impact reaction outcomes, aiding chemists in designing more efficient synthetic routes and optimizing reaction conditions.
Difficulties in Constructing and Employing Reaction Datasets:
Although large-scale reaction datasets provide substantial advantages, they also present their distinct obstacles. The problems encompass data incompleteness and bias, assuring precision in mechanistic annotations, computational hurdles in managing extensive datasets, and integration with established chemical databases.
Insufficient data and prejudice:
Data incompleteness is a significant obstacle when it comes to constructing and utilizing extensive reaction datasets. Certain datasets may lack information on certain mechanistic stages or intermediate states, and not all reactions receive complete recording. This might result in biases in prediction models, potentially compromising their performance when dealing with reactions that deviate greatly from those included in the dataset.
Data incompleteness might stem from various origins. For instance, the literature may document certain reactions without providing comprehensive information about the reaction conditions or the identification of intermediate substances. Alternatively, the data may be incomplete due to the challenging nature of experimentally observing or characterizing some mechanistic steps. The absence of sufficient data can constrain the capacity of predictive models to precisely forecast the results of novel reactions.
Another notable obstacle to reaction datasets is bias. Bias may arise when the data inside a dataset fails to encompass the complete spectrum of potential responses. For instance, if a dataset exhibits an imbalanced distribution of reactions involving specific types of reactants or circumstances, the resulting predictive models may display a bias toward these reactions. When the models apply to reactions that differ from those in the dataset, this bias could lead to inaccurate predictions.
Ensuring Precision in Mechanistic Annotations:
Ensuring the quality of mechanistic annotations is a significant difficulty when it comes to developing and utilizing response databases. Mechanistic annotations are essential for understanding the progression of a reaction and forecasting the results of analogous processes. Nevertheless, the process of annotating mechanistic processes presents difficulties, especially when dealing with intricate reactions that consist of many steps or involve uncommon intermediates.
An obstacle to achieving precision lies in the intricate nature of certain reaction systems presents an obstacle to achieving precision. Sometimes, we fail to understand the precise mechanism, or the reaction may involve multiple competing pathways. The intricate nature of the task can present challenges in appropriately annotating the mechanical processes, hence increasing the likelihood of errors in the dataset.
Another challenge is the dependence on computational models for forecasting mechanistic pathways. Although these models can offer significant insights, their accuracy is not always reliable, especially for reactions that involve uncommon or intricate systems. To ensure accuracy in mechanistic annotations, it is necessary to meticulously validate computational predictions using experimental data and expert knowledge. Mechanistic Pathways of Organic Reactions:
There are challenges in processing large datasets with computers:
The vast amount of data in extensive reaction datasets poses significant computing challenges. These datasets may consist of millions of distinct responses, each characterized by its unique factors and conditions. To effectively store, process, and analyze this data, it is necessary to have a strong computational infrastructure and advanced algorithms.
Data storage is a significant computational obstacle. Extensive reaction datasets necessitate significant storage capacity, especially when they encompass comprehensive details regarding reactants, products, intermediates, and reaction conditions. A substantial difficulty lies in ensuring the effective storage and simple accessibility and querying of this data by academics.
Data processing poses another computational issue. Examining extensive datasets necessitates substantial computer capacity, especially when employing machine learning methods to detect patterns and correlations. It is crucial to ensure that these algorithms can swiftly and accurately process the data to maximize the usefulness of the information.
Incorporation into Preexisting Chemical Databases:
Another major problem is the integration of extensive reaction datasets with existing chemical databases. Chemical databases have extensive data on molecules, reactions, and chemical properties. We can gain valuable insights by merging this knowledge with reaction datasets. Nevertheless, this connection necessitates meticulous coordination and consistency across diverse platforms.
A significant obstacle in the process of integration is ensuring compatibility between different data formats. Various databases may employ distinct forms to depict chemical reactions, such as SMILES, InChI, or reaction SMIRKS. To achieve seamless data integration and cross-referencing from multiple databases, it is necessary to standardize data formats and meticulously curate the data.
Another obstacle that arises is the issue of accessing and sharing data. Private ownership of certain chemical databases requires subscriptions or permits for entry. Facilitating the integration of data from these databases with open-access reaction datasets necessitates meticulous discussion and collaboration between database suppliers and researchers.
Notable Large-Scale Reaction Datasets:
Numerous extensive reaction databases have become crucial in organic chemistry research. The files offer researchers extensive access to a wealth of information regarding chemical processes, encompassing intricate mechanistic pathways, reaction circumstances, and outcomes. The subsequent sections offer a synopsis of many prominent and extensive response datasets.
Reaxys is a database:
Reaxys is a widely used collection of organic chemistry reaction data. The platform offers users access to an extensive compilation of chemical reactions, accompanied by comprehensive data on the substances involved, including reactants, products, intermediates, and the conditions under which the reactions occur. Reaxys is highly beneficial for researchers who aim to investigate unexplored chemical territory or develop novel synthetic pathways.
Reaxys’s comprehensive inclusion of empirical data is an essential characteristic. The dataset comprises reactions from diverse sources, encompassing published literature, patents, and experimental findings. Reaxys provides comprehensive coverage, making it a helpful resource for academics seeking to comprehend specific reactions or delve into new areas of chemistry.
Reaxys offers sophisticated search and analysis capabilities that enable researchers to promptly discover pertinent reactions and investigate patterns in reaction behavior. These tools encompass reaction similarity searches, enabling researchers to identify reactions that bear resemblance to a particular reaction, relying on structural or mechanistic similarities.
PubChem is a chemical database:
PubChem is a popular and extensively used chemical reaction database, providing unrestricted access to a large assortment of reactions and associated data. PubChem is highly advantageous for academic researchers due to its ability to facilitate extensive use and collaboration without requiring costly subscriptions or licenses.
PubChem is notable for its emphasis on providing unrestricted access to data. The dataset is accessible to academics globally, serving as a vital resource for individuals who lack access to exclusive datasets. PubChem offers an extensive array of data types, such as chemical structures, bioactivity data, and reaction data, which makes it a useful resource for researchers in diverse domains.
PubChem offers sophisticated search and analytical capabilities, enabling academics to thoroughly investigate the dataset. The tools provided include structure searches, enabling researchers to locate reactions involving certain chemical structures, and bioactivity searches, enabling researchers to investigate the biological activity of various substances.
Datasets tailored to specific industries:
Specialized datasets, developed specifically in the areas of pharmaceuticals and materials research, complement comprehensive datasets like Reaxys and PubChem. Researchers frequently use these datasets, which often contain confidential information, to drive advancements in pharmaceutical research and the creation of novel substances.
An instance of an industry-specific dataset is the GDB (Generated DataBase) series, which encompasses extensive compilations of diminutive organic compounds produced by computer techniques. Pharmaceutical companies use this information to investigate unexplored chemical spaces and discover potential medication opportunities.
Another instance is the Materials Project, which offers an extensive collection of material attributes, encompassing information on crystal structures, electrical properties, and reaction thermodynamics. Researchers in materials science utilize this dataset to facilitate the development of novel materials possessing certain characteristics, such as enhanced conductivity or reduced thermal expansion.
Artificial intelligence (AI) and machine learning play a crucial role in analyzing reaction datasets:
The incorporation of artificial intelligence (AI) and machine learning (ML) into extensive reaction datasets has brought about a significant transformation in the domain of organic chemistry. Artificial intelligence (AI) and machine learning algorithms can evaluate large volumes of data, enabling the identification of patterns and connections that may not be readily observable to human researchers. We can use these observations to predict reaction outcomes, improve the reaction environment, and even suggest new pathways for reaction mechanisms.
We use machine learning algorithms to forecast reaction outcomes:
A prominent use of artificial intelligence in the field of organic chemistry is employing machine learning algorithms to forecast the results of chemical reactions. These algorithms use extensive datasets of documented reactions to identify patterns and correlations, which they then use to predict the outcomes of untested reactions.
For instance, a machine learning program could examine a dataset of reactions involving a certain type of reactant and detect recurring patterns in the reaction conditions that result in significant yields or selectivity. Afterward, chemists can use this data to predict the outcome of a new reaction with a similar starting material, aiding them in creating more efficient and targeted synthetic pathways.
We can also employ machine learning methods to forecast the development of secondary products or the incidence of secondary reactions. By analyzing data from previous experiments, these algorithms can identify the parameters associated with the production of undesirable by-products. Furthermore, they can propose adjustments to the reaction conditions to decrease the production of these undesired substances.
Artificial intelligence (AI) is in the process of understanding and explaining mechanistic pathways:
We are using AI to clarify the molecular pathways involved in chemical reactions. AI algorithms can use huge amounts of reaction data to look at and suggest possible mechanisms for reactions with unclear pathways. They can also suggest ways to improve existing mechanisms so that they fit the data better.
For instance, an AI algorithm may examine a dataset of reactions about a certain type of bond creation and detect recurring patterns in the sequential stages that result in the establishment of a successful bond. Afterward, chemists can use this data to suggest a possible mechanism for a new reaction involving a similar chemical bond, which will enhance their understanding of the reaction’s progression and allow them to optimize conditions for higher yields.
Artificial intelligence (AI) is especially beneficial in situations where the precise process of a reaction is uncertain or involves numerous competing routes. Artificial intelligence (AI) algorithms analyze extensive datasets of reaction data to identify the most probable routes and suggest potential intermediates or transition states that may not have been considered previously.
Case Studies: Effective Implementations of Artificial Intelligence in Reaction Prediction
Multiple case studies demonstrate the effective utilization of AI in predicting reactions and clarifying mechanistic pathways. These case studies show how AI can change organic chemistry by giving chemists new ways to look at how reactions work and helping them come up with more efficient and selective ways to make chemicals.
An example is the use of artificial intelligence to forecast the results of Suzuki-Miyaura cross-coupling reactions. A machine learning system was used to look at a large set of cross-coupling reactions and find links between the conditions of the reactions and good results in terms of both high yields and selectivity. They then used the method to predict the outcomes of new cross-coupling processes with remarkable precision. They used this data to create more efficient synthetic pathways, which reduced the need for laboratory empirical investigation.
Another case study examines the use of AI to clarify a complex reaction’s step-by-step process. Researchers employed an AI algorithm to scrutinize an extensive dataset of reaction data and ascertain the most probable routes for the creation of the intended product. The program suggested a previously undiscovered intermediate, which experimentation later confirmed. This discovery enhanced our comprehension of the reaction process and facilitated the creation of more effective synthetic pathways.
We must consider ethical and legal factors:
The use of extensive reaction databases also raises significant ethical and legal concerns. These factors include concerns about the protection of data privacy and intellectual property, the importance of promoting open science and data sharing, and adherence to regulations in chemical data administration.
Data Privacy and Intellectual Property Concerns:
Data privacy is a significant ethical concern when working with extensive reaction datasets, especially when dealing with private or sensitive information. Reaction datasets are frequently composed of experimental findings, published scientific literature, and computational forecasts. Note that intellectual property rights or other legal constraints may govern certain portions of these datasets.
Safeguarding private information is crucial to preventing legal entanglements and upholding the integrity of the research. Researchers must adhere to intellectual property regulations to ensure responsible data usage and prevent the unauthorized disclosure of sensitive information.
Data privacy is a crucial factor to consider while using reaction datasets, along with worries about intellectual property. When using data, researchers must ensure the protection of personal or sensitive information, as well as adherence to relevant privacy laws and regulations.
The importance of open science and data sharing:
Another crucial ethical aspect is the significance of doing open science and sharing data. Although certain response datasets are exclusive and necessitate subscriptions or licenses for accessibility, numerous members of the scientific community push for open access to data to foster collaboration and expedite scientific breakthroughs.
Open science and data sharing enables global researchers to access and utilize reaction datasets, regardless of their institutional affiliation or financial means. Providing unrestricted access to data can facilitate novel discoveries, promote cooperation, and expedite scientific advancements.
Nevertheless, the profession is still struggling to find a solution to the dilemma of maintaining open access while safeguarding intellectual property and proprietary information. It is critical to uphold research integrity by responsibly and ethically sharing data according to legal norms.
Regulatory Compliance in Chemical Data Management:
Ensuring adherence to regulations is another crucial factor to take into account when utilizing extensive amounts of reaction data. In high-stakes businesses such as pharmaceuticals, it is crucial to adhere to all applicable standards for data collection, storage, and utilization. This is necessary to prevent legal difficulties and maintain the integrity of research.
In the pharmaceutical sector, strict adherence to requirements from authorities such as the Food and Drug Administration (FDA) or the European Medicines Agency (EMA) is necessary to effectively handle data related to chemical reactions and drug candidates. It is crucial to ensure that reaction databases are precise, comprehensive, and under these regulations to successfully introduce new medications to the market.
Researchers must ensure that their utilization of reaction datasets adheres to both industry-specific restrictions and general data protection rules, such as the General Data Protection Regulation (GDPR) in the European Union. These regulations mandate the careful handling of personal and sensitive data, as well as the requirement to inform data subjects about their data usage.
Prospects for Advancements in Reaction Dataset Development:
The subject of reaction dataset development is undergoing tremendous evolution, with numerous promising ideas emerging. These developments encompass the growing significance of quantum computing in analyzing mechanistic pathways, the collaborative endeavors among academia, industry, and government, and the incorporation of novel data formats into response databases.
Emerging Patterns for Gathering and Annotating Data:
A notable trend in the development of reaction datasets is the growing complexity of data gathering and annotation methods. Recent progress in high-throughput screening, automated testing, and computer modeling has enabled the collection and annotation of response data on an unprecedented scale.
Automated experimentation platforms have advanced to the point where they can conduct thousands of reactions in a single day, generating large quantities of data for incorporation into reaction datasets. These platforms can additionally streamline the process of annotating mechanistic stages, minimizing the risk of human error and guaranteeing the accuracy and comprehensiveness of the data.
Furthermore, alongside progress in data collection, there are notable advancements in data annotation. Researchers are now developing novel techniques and algorithms to automatically label mechanistic processes, detect intermediates, and forecast reaction outcomes. These methods enable the creation of more intricate and precise reaction databases, equipping researchers with the necessary information to devise more effective and specific synthetic pathways.
The role of quantum computing in mechanistic pathway analysis is significant:
Quantum computing is playing an increasingly important role in the analysis of mechanistic pathways, which is an intriguing trend in the creation of reaction datasets. Quantum computers can simulate intricate chemical systems with unparalleled precision, creating new opportunities for understanding and forecasting chemical events.
Quantum computers excel at mimicking the behavior of molecules and reactions at the quantum level, a task that regular computers find challenging. Quantum computers can simulate the quantum states of molecules, allowing them to offer valuable insights into reaction mechanisms that are challenging or unattainable for classical computers.
Quantum computers have the potential to model the transition stages of intricate reactions, offering precise insights into the energy barriers and intermediates that are part of the process. We could then use this data to improve mechanistic pathways and the accuracy of reaction forecasts.
Although quantum computing is now in its nascent phase, it holds immense potential for applications in mechanistic pathway research. We anticipate that the power and accessibility of quantum computers will significantly influence the development of extensive reaction datasets.
Collaborative Endeavors: Academic, Industrial, and Government Initiatives
Academics, industry, and government will greatly influence the future of reaction dataset development through collaborative endeavors. These partnerships can facilitate the establishment of uniform procedures for gathering and labeling data, guaranteeing that datasets are thorough, precise, and easily available to a large audience.
Government programs such as the National Institutes of Health (NIH) Data Commons are actively involved in establishing a collaborative framework for biomedical data, which includes response datasets. These programs seek to foster collaboration and facilitate the exchange of data among many disciplines and organizations, thereby expediting the rate of scientific breakthroughs.
Collaborations between academics and industry are playing a crucial role in the development of response datasets, in addition to government initiatives. Academic researchers frequently lead the way in creating novel techniques for gathering and annotating data, while industry partners contribute the necessary resources and experience to apply these techniques to extensive datasets.
Through collaboration, the academic, industrial, and governmental sectors can generate more extensive and precise reaction databases, equipping researchers with the necessary resources to propel innovation and exploration in the field of organic chemistry.
In conclusion:
Mechanistic Pathways of Organic Reactions, The use of extensive reaction datasets is revolutionizing organic chemistry, providing an unparalleled understanding of mechanistic pathways and facilitating novel approaches to research and development. These datasets offer researchers the necessary information to formulate experiments, forecast reaction results, and investigate uncharted domains of chemical space.
We expect that as the field of making reaction datasets grows, more advanced datasets will appear that include a wider range of data sources, such as real-time experimental results and computational predictions. The incorporation of artificial intelligence (AI) and machine learning (ML) with these datasets holds the potential to unleash further opportunities, rendering it a thrilling period to engage in this domain.
The capability to get and examine extensive reaction data is increasingly becoming a crucial instrument for chemists globally, whether it be in drug discovery, materials science, or fundamental research. As large-scale reaction databases continue to expand and develop, they will surely have a progressively significant impact on shaping the future of chemistry.
Frequently Asked Questions:
1). What is the mechanistic pathways of organic Reactions?
A mechanistic pathway is a comprehensive and sequential account of the processes that take place during a chemical reaction, illustrating the conversion of reactants into products.
2). How are large-scale reaction datasets used in drug discovery?
These datasets help researchers find possible synthetic pathways for new drug candidates, guess how reactions will go, and find the best reaction settings to increase yields and selectivity.
3). What difficulties arise while trying to ensure precision in reaction datasets?
The obstacles encompass issues such as inadequate data, biases within the data, the intricacy of mechanistic annotations, and the computational difficulties associated with managing extensive datasets.
4). What role does AI play in improving our understanding of organic reactions?
Artificial intelligence can analyze large datasets to predict reaction outcomes, suggest potential pathways for those reactions, and identify patterns in reaction behavior that can direct the creation of new synthetic routes.
5). Do researchers have access to extensive, publicly accessible collections of chemical reaction data?
Indeed, databases such as PubChem provide unrestricted access to extensive collections of reaction statistics, allowing researchers from around the world to obtain and use the data for their research and collaborative efforts.
For more chemistry blogs, visit chemistry Master