Table of Contents
Overview of Multi-Target Ligands (MTLs):
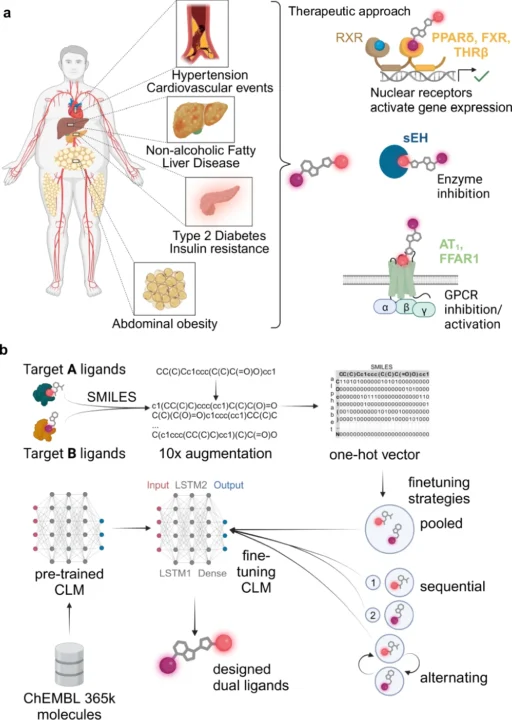
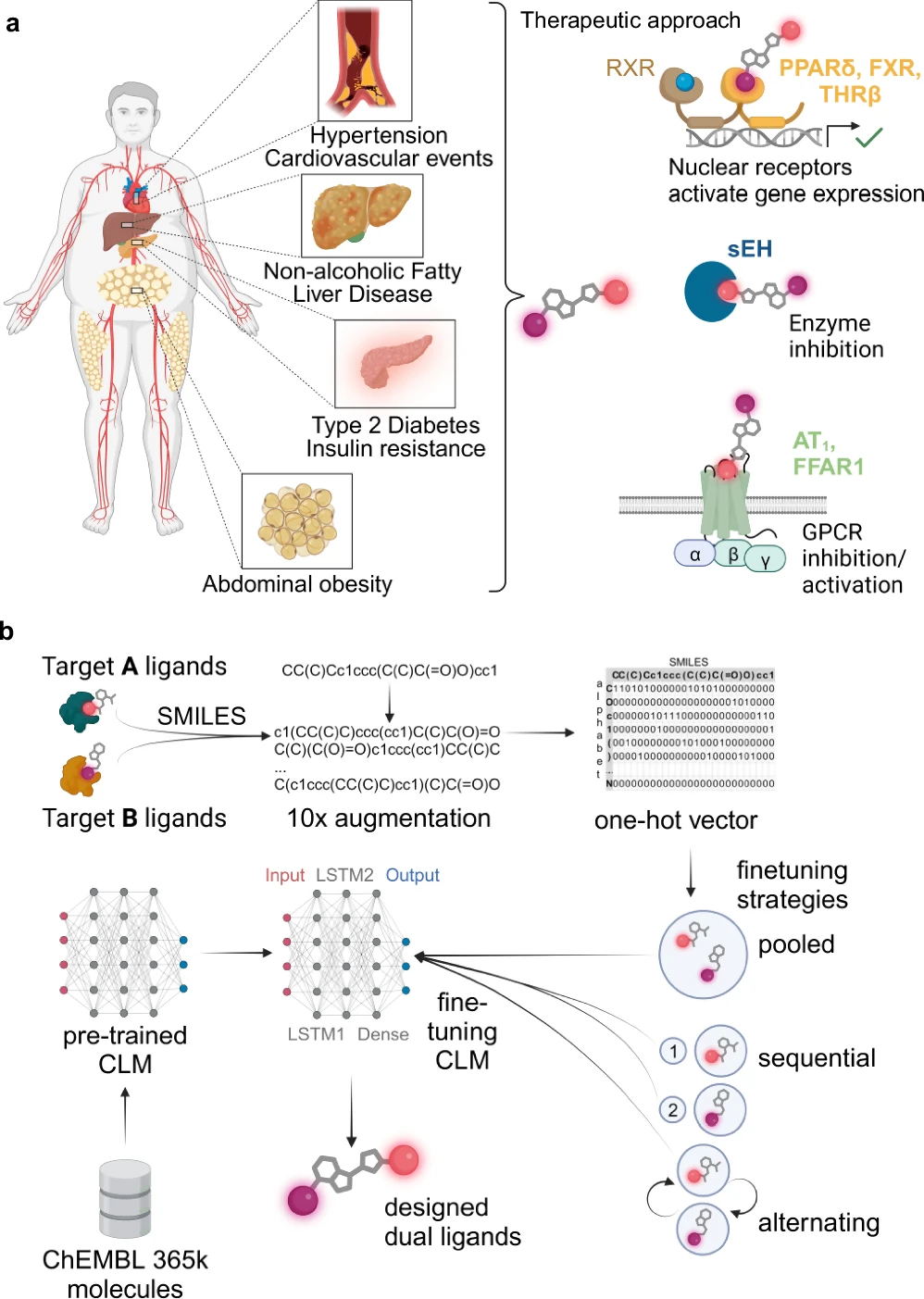
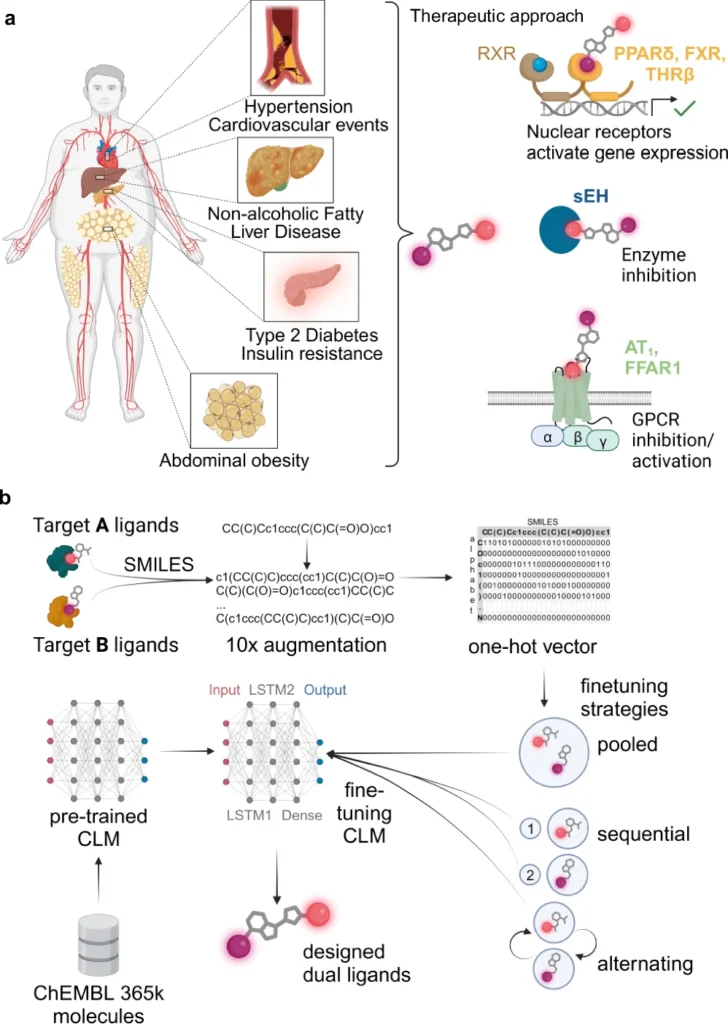
The design of compounds that may engage with several biological targets, referred to as multi-target ligands (MTLs), is becoming prominent in drug discovery. Ligands are chemicals that attach to receptors and trigger a biological reaction. Historically, medication development concentrated on formulating compounds that target a certain receptor or pathway. Nevertheless, numerous diseases, particularly intricate ones such as cancer, Alzheimer’s, and diabetes, engage multiple biological targets. Multi-target ligands, engineered to influence many pathways concurrently, provide a more comprehensive therapeutic strategy.
Nonetheless, the design of Multi-Target Ligands (MTLs) presents significant challenges. The challenge of identifying a chemical that can effectively bind to several targets while minimizing off-target effects is considerable. Conventional drug discovery techniques, including high-throughput screening and empirical experimentation, are laborious and expensive. Let’s introduce deep learning as a transformative force that can automate the design of Multi-Target Ligands (MTLs). Metabolic syndrome and application of a chemical language model for multi-target de novo design.
The role of deep learning in pharmaceutical discovery:
Artificial intelligence (AI) has transformed numerous industries and is now redefining pharmaceutical research. Deep learning, a branch of artificial intelligence, emulates the neural networks of the human brain, enabling the analysis of extensive information and the identification of patterns. By analyzing extensive chemical and biological datasets, deep learning has demonstrated significant efficacy in drug development, revealing promising therapeutic candidates that conventional approaches may have missed.
Generative modeling is one of the most exhilarating applications of deep learning in drug discovery. This method enables researchers to synthesize novel chemical compounds by analyzing existing data. In the realm of Multi-Target Ligands (MTLs), generative deep learning models can suggest innovative compounds capable of binding to many targets, hence accelerating the drug discovery process considerably.
An Exposition on Generative Deep Learning:
Generative deep learning refers to the use of algorithms to generate new data points from existing datasets. It diverges from conventional machine learning, which predominantly emphasizes categorization or regression problems (such as forecasting a drug’s efficacy based on input characteristics). In generative deep learning, the objective is to synthesize novel chemical compounds that are not yet in existence but may serve as potential therapeutic candidates.
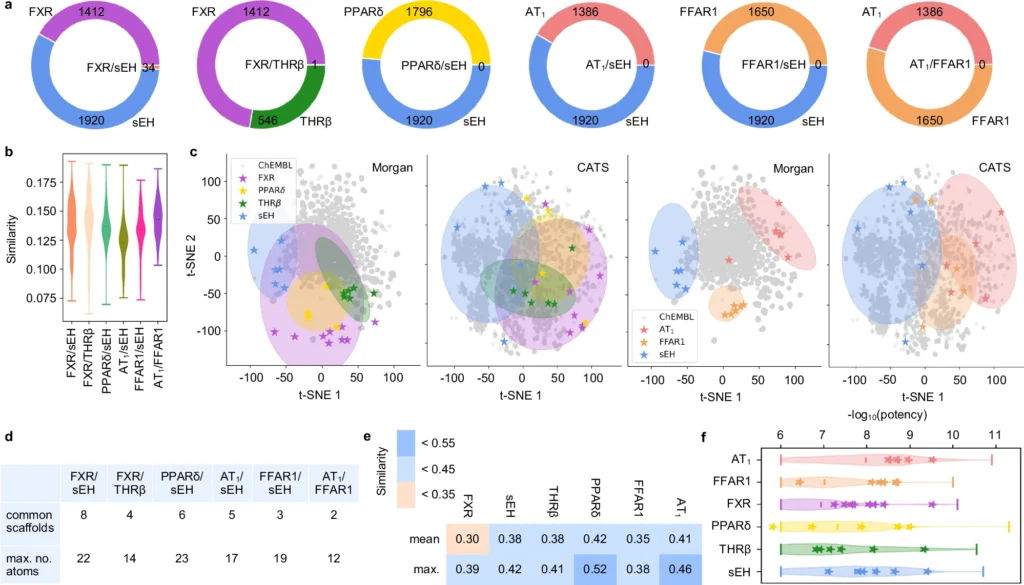
This methodology is particularly advantageous in chemistry and pharmaceutical creation, where scientists are always seeking novel compounds with distinct characteristics. Generative models, like Variational Autoencoders (VAEs) and Generative Adversarial Networks (GANs), let scientists look into chemical spaces that are much bigger than what we already know. Analysis of known ligands for targets of interest.
Comprehending Multi-Target Ligands Design:
Developing multi-target ligands (MTLs) is more intricate than formulating single-target pharmaceuticals. The goal is to design a chemical that can bind to multiple distinct biological receptors with outstanding selectivity and efficiency. Multi-target ligands need to find a delicate balance: they need to interact strongly with many targets while minimizing interactions with other targets, which can have negative effects or lower the effectiveness of a drug.
Often, the targets may be interconnected yet architecturally distinct, introducing an additional layer of complexity. This necessitates precision which is challenging to get with conventional experimental techniques, rendering deep learning an appealing alternative. Fine-tuning of chemical language models (CLM) for multi-target design.
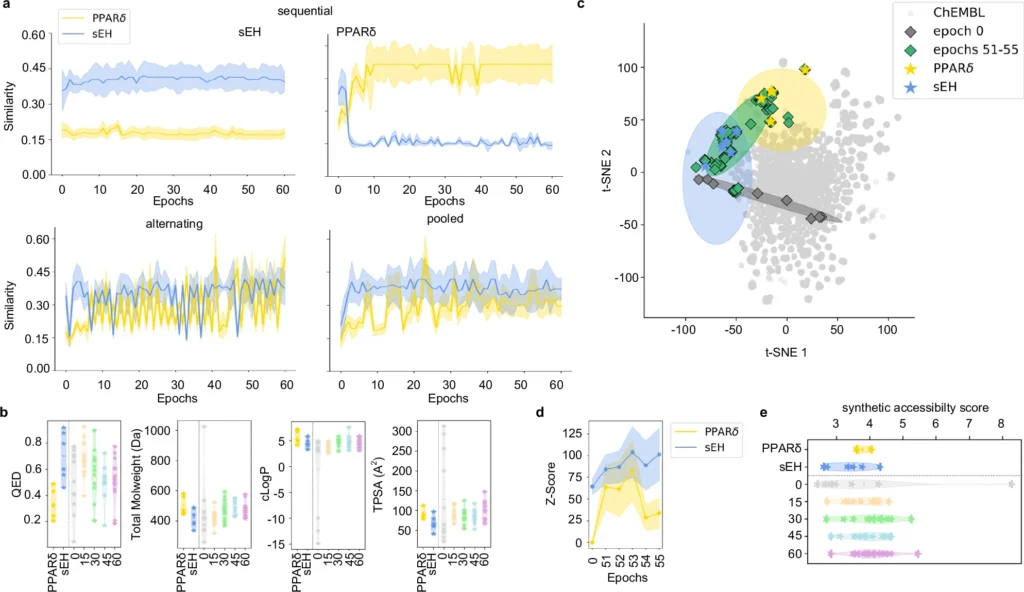
Obstacles in Conventional Ligand Design:
Before the advent of AI, ligand design was an arduous endeavor. Researchers utilized experimental methodologies such as high-throughput screening, which entailed evaluating hundreds or even millions of molecules to identify one with the desired qualities. This method was both time-consuming and costly, as well as riddled with inefficiencies. Numerous attractive candidates may fail in the later stages of research, resulting in significant costs in both time and resources for pharmaceutical corporations.
Computational techniques, including molecular docking and quantitative structure-activity relationship (QSAR) models, enhanced this process. Nevertheless, these methodologies exhibited constraints, especially in the context of multi-target design, which necessitates the equilibrium of interactions across many biological targets.
Generative Deep Learning for Ligand Synthesis:
Generative deep learning automates multi-target ligand design by creating unique compounds that fulfill numerous objectives. Deep learning methods acquire knowledge from extensive datasets of recognized ligands and their interactions with biological targets. These models can suggest novel compounds that may attach to various targets with significant affinity when trained.
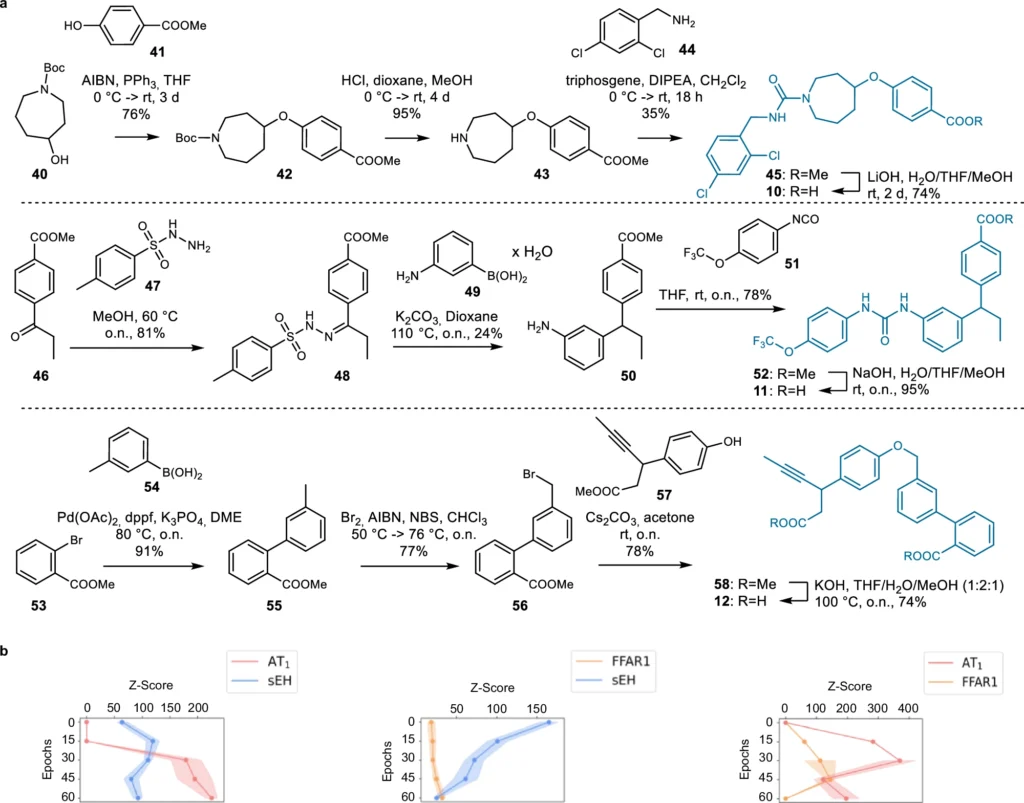
These AI models function by discerning the fundamental patterns in chemical data, enabling them to produce innovative structures. Using deep learning for ligand design has many benefits, such as being fast, effective, and able to look into chemical areas that other methods might miss. Synthesis of the dual ligand candidates 1–9 designed by the chemical language model (CLM).
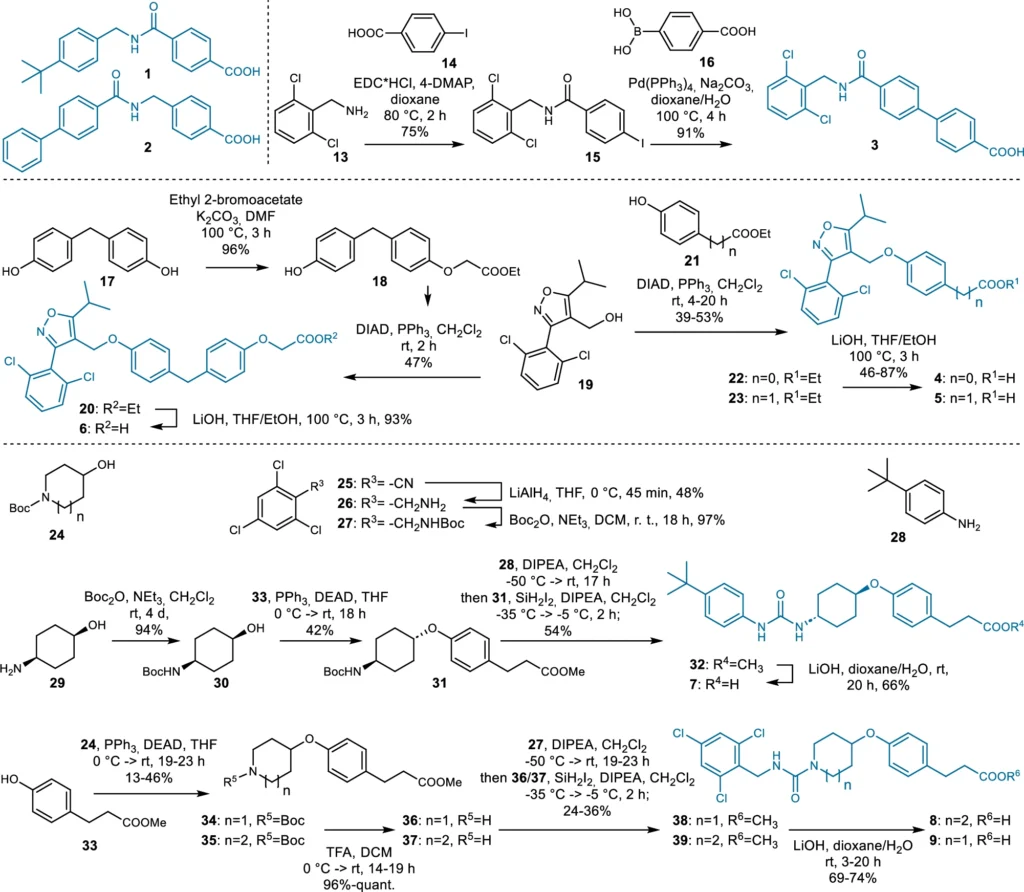
Instances of Generative Models in Pharmaceutical Design:
The design of ligands uses a variety of generative models:
Variational Autoencoders (VAEs): We frequently use variational autoencoders (VAEs) to synthesize novel compounds by acquiring the probability distribution of chemical data.
Generative Adversarial Networks (GANs): Generative Adversarial Networks (GANs) consist of two neural networks: one that generates molecules and another that assesses them, progressively enhancing the realism of the structures created.
Recurrent Neural Networks (RNNs): We frequently use recurrent neural networks (RNNs) to generate molecular sequences, which are particularly advantageous for the synthesis of large, intricate compounds.
Training deep learning models for ligand design:
The efficacy of a generative deep learning model in ligand creation is highly dependent on the quality of the data used. For training these models, extensive databases of chemical substances and their documented interactions are critical. The processing stages, including the normalization of chemical structures and the assurance of data consistency, are essential. The models undergo several training steps to learn to produce realistic and practical molecular architectures. Features of dual ligands designed by the chemical language models (CLM).
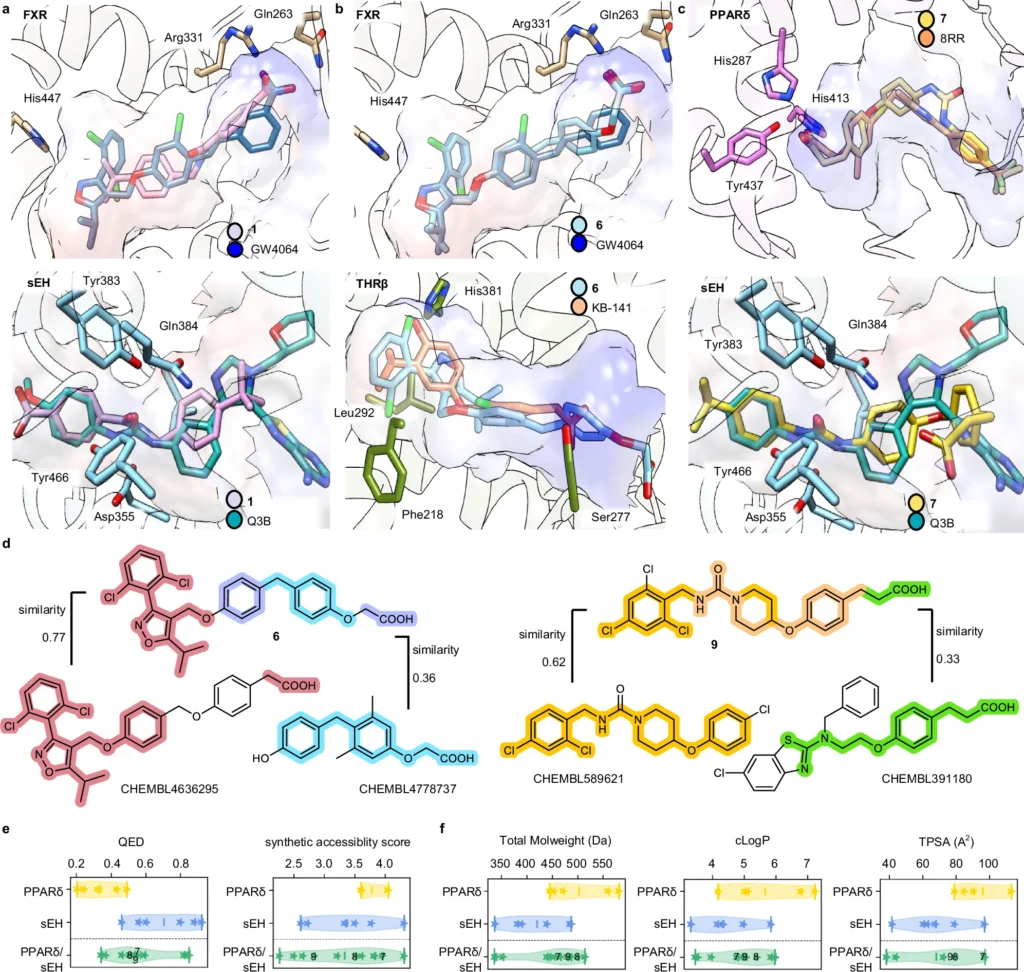
Case Studies of Multi-Target Ligands Designed by AI:
Numerous case studies underscore the efficacy of AI-engineered Multi-Target Ligands (MTLs). Artificially intelligent multi-target ligands have shown promise in the treatment of neurodegenerative diseases like Alzheimer’s, which affect many biological pathways. AI models have facilitated new pathways in drug discovery by creating compounds that concurrently target amyloid plaques and tau proteins.
The Opportunities for Pharmaceutical Discovery Using Generative Models:
Generative deep learning is poised to revolutionize the future of drug development. As AI models advance, they will increasingly anticipate with enhanced precision the interactions of novel ligands with various biological targets. This accelerates the discovery process and lowers the costs associated with unsuccessful medication trials. Validation and comparison of the dual ligand design approach.
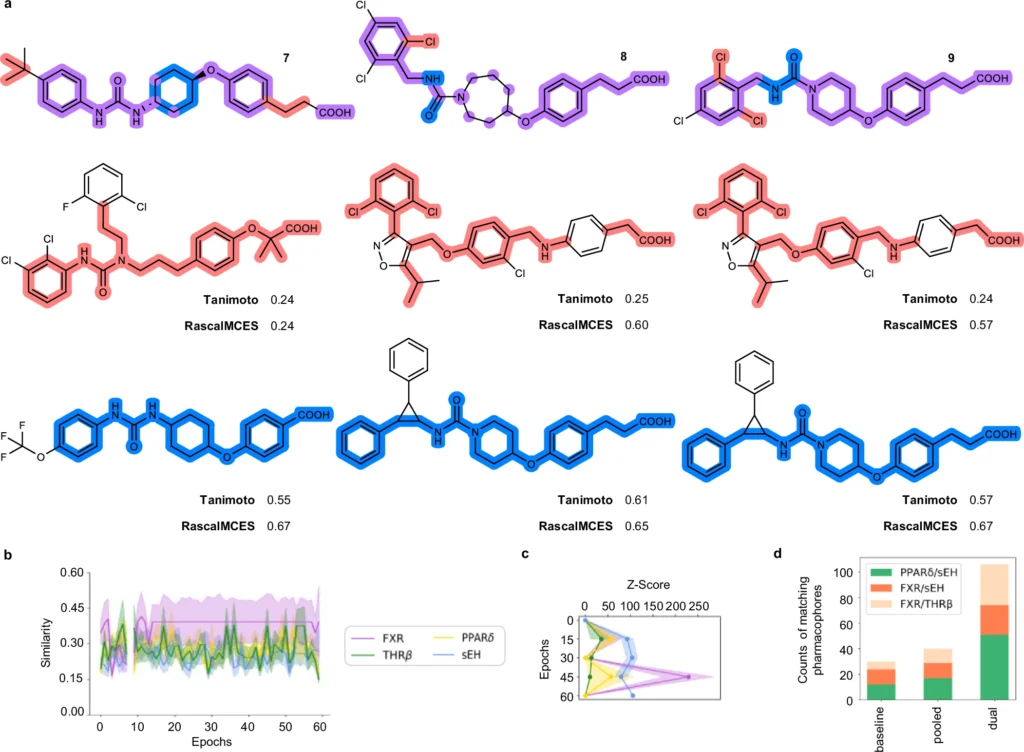
Advantages of AI-Generated Machine Translation Lists:
One of the primary benefits of using AI in drug design is the speed with which it can provide viable candidates. We can now accomplish what used to require years of laboratory research in a significantly shorter timeframe. AI facilitates the exploration of uncharted chemical regions, resulting in the identification of totally new classes of drugs.
The Limitations of Generative Deep Learning in Multi-Task Learning Design:
Notwithstanding their potential, generative deep learning models possess inherent limits. The bias that may emerge from training data is a problem. If the training set lacks sufficient diversity, the model may produce molecules that closely resemble existing ones, constraining creativity. Moreover, although AI can synthesize novel chemicals, comprehending the mechanisms behind a molecule’s efficacy frequently necessitates supplementary interpretability tools.
Enhancing Generative Deep Learning Models for Ligand Design:
Researchers are persistently enhancing generative deep learning models. Enhancing the quality and diversity of the data used for training these models is one area of emphasis. Another significant improvement is the creation of superior algorithms capable of balancing several objectives, including affinity for various targets and avoidance of off-target encounters. Designed dual ligand candidates for AT1/sEH (10), FFAR1/sEH (11), and FFAR1/AT1 (12) from chemical language models.
Regulatory and Ethical Considerations:
Like any AI-driven technology, regulatory and ethical issues require attention. Regulatory agencies must adjust to the surge of AI-generated pharmaceuticals, guaranteeing their safety and efficacy. Ethical considerations, including the transparency of AI algorithms and the potential for misuse, must be considered.
Final Assessment:
Generative deep learning signifies a novel advancement in drug discovery, especially in the design of multi-target ligands. Its capacity to automate design, investigate novel chemical spaces, and optimize for various targets presents a groundbreaking method for addressing intricate disorders. As artificial intelligence advances, the capacity to identify novel, life-saving pharmaceuticals will increase.
Frequently Asked Questions:
1). What are multi-target ligands?
Multi-target ligands are compounds engineered to engage with various biological targets, providing a more holistic therapeutic strategy, particularly for intricate disorders.
2). In what ways does deep learning contribute to medication design?
Deep learning facilitates the analysis of extensive datasets to identify patterns and produce novel molecules that may serve as potential therapeutic candidates.
3). What are the primary categories of generative models employed in drug design?
The primary categories of generative models include Variational Autoencoders (VAEs), Generative Adversarial Networks (GANs), and Recurrent Neural Networks (RNNs).
4). What challenges do conventional ligand designs face?
Conventional ligand design is laborious, costly, and frequently inadequate in addressing the intricacies of multi-target interactions.
5). What are the AI constraints in ligand design?
Limitations encompass biases in training data, difficulties in comprehending the model’s thinking, and existing constraints in tackling very intricate biological systems.
For more chemistry blogs, visit chemistry Master